
Thermodynamics in Brief
Thermodynamics is that part of science which is concerned with the conditions that material systems may assume and the changes in conditions that may occur either spontaneously or as a result of interactions between systems. The word "thermodynamics" was derived from the Greek words thermé (heat) and dynamics (force). The formulation of the First and Second Law of Thermodynamics by the German scientist Rudolf Julius Clausius in 1850 lay the foundation for what is now called "classical" or "equilibrium thermodynamics." More recently, thermodynamics has been extended to include nonequilibrium states.

The description of physical phenomena is based on the concept of the state of a system and the changes that occur spontaneously or through interaction with other systems. The term system means any identifiable collection of matter that can be separated from everything else by a well-defined surface. Examples for thermodynamic systems are the water molecules in a container or, much more complex, a complete process plant. If the system boundaries permit the exchange of heat and work, but not of physical matter, the system is termed Closed System, as compared to the Open System, where mass transfer may occur. At any time, a system is in a condition called "state" which encompasses all that can be said about the results of any measurements or observations that can be performed on the system at that time, it is necessary to distinguish between quantities which depend on the path between states (such as heat and work) and those which depend solely on the state (such as temperature, internal energy or entropy).
A system is in equilibrium if there is no tendency for a change in state to occur, i.e. if all forces are in exact balance. The number of independent intensive (mass-independent) properties that must be arbitrarily fixed to establish the state of a system is named the degree of freedom. It can be calculated according to Gibbs
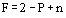
with P being the number of phases and n being the number of chemical species present.
A process is a change in the system from an initial state to a final state. Processes can be divided into two types, namely reversible processes which can be reversed at any time so that the system and the surroundings are returned to their original condition and irreversible processes in which the reversal cannot be carried out without leaving some change in the system or the surroundings. Reversible processes are ideal processes in the absence of friction and finite temperature differences.
Four laws form the foundation of thermodynamics, even though the First and the Second Laws are considered the most important.
The Zeroth Law is the basis for the measurement of temperature. It states that "two bodies which are in thermal equilibrium with a third body are in thermal equilibrium with each other."
The First Law of Thermodynamics formally states that "while energy assumes many forms, the total quantity of energy is constant. When energy disappears in one form it appears simultaneously in other forms." This may be written as

In traditional processes, changes may occur in internal energy, in potential energy and kinetic energy. The recognition of heat and work as energy in transit leads to the following formulation of the First Law

where Q is heat supplied and W work done.
Closed Systems often undergo processes with no changes in kinetic or potential energy, hence

Far more important industrially are processes which involve the steady-state flow of fluids through equipment, see, for example, Figure 1.
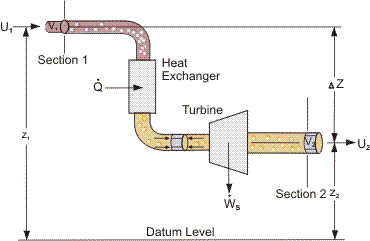
For these Open Systems, a more general expression of the First Law is used. The total work exchanged between the system and its surroundings consists of the shaft work Ws and the flow work due to pressure forces as the fluid enters and leaves the system.
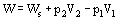
where p is pressure and V volume.
With the definition of enthalpy, Equations (3) and (5) can be combined to give

where H is enthalpy, M is mass, u is velocity and z height. A cycle process is a process in which the initial and final states are identical. By virtue of the First Law, the work of an adiabatic cycle (Q = 0) must be zero.
The microscopic disorder of a system is described by a system property called Entropy. The Second Law of Thermodynamics states that whenever a process occurs, the entropy of all systems involved in the process must either increase or, if the process is reversible, remain constant. "As a result, it is impossible to construct a machine operating in a cyclic manner which is able to convey heat from one reservoir at a lower temperature to one at higher temperature and produce no other effect on any other part of the surrounding." This formulation of the Second Law has been attributed to Clausius. The state function entropys is defined as

where dQrev is the reversible heat input and T the absolute temperature, and the Second Law can then be written as

where subscripts tot, sys and sur refer to total, system and surroundings.
The Second Law of Thermodynamics has many far-reaching implications, for example, the criterion that for a system in stable equilibrium, the entropy of the system must be at its maximum for fixed values of energy, number of particles and physical constraints.
Nernst's Law is frequently named the Third Law of Thermodynamics. It states that "the entropy of a thermodynamic system approaches zero for T → 0 K." This implies that the specific heat capacity of all substances approaches zero, too. As a result, the entropy at any temperature can be calculated from

where N is the number of moles, cp the molar specific heat and the subscript ph refers to phase change.
The Exergy (sometimes also called availability) is a measure of the maximum useful work that can be produced (or the minimum work required) by a system interacting with the environment. It can be achieved by operating reversibly within the system and transferring heat reversibly. For a closed system
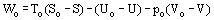
and for an open system

with subscript "o" denoting reference conditions which are usually taken as 25°C and 1 atm. If the process does not end at ambient conditions, the ideal work is

for a closed system and

for an open system. Obviously, this ideal work does not exist. It only serves as a basis for thermodynamic calculations. The lost work is the difference between the ideal work and the work actually done by the process

For closed processes, the following can be derived

while for the practically more important flow processes

A thermodynamic efficiency can be defined as:
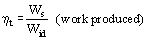

The second law and the associated principle of increase of entropy provide the basis for the analysis of equilibrium problems. For a compressible substance undergoing a general process in which heat and work may be exchanged with the system

Assuming that temperature and pressure of the surrounding are constant

and that the system and surrounding are in equilibrium with respect to temperature and pressure
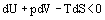
where all properties refer to the system. Equation (21) may be more conveniently written in terms of Helmholtz and Gibbs Functions. The Helmholtz energy H is defined by
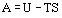
and Gibbs energy G by
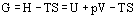
and hence


For constant pressure and temperature, the Helmholtz and Gibbs energies must decrease and seek minimum values. Once these minima are attained, equilibrium conditions will have been reached with no further change in A and G. The important criterion for equilibrium is therefore
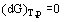
for a single-component system and

for a multicomponent system, where Ni and 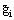 are the number of moles and the molar Gibbs energy of each constituent.
are the number of moles and the molar Gibbs energy of each constituent.
If we consider processes without change in composition, the Fundamental Equations of Thermodynamics can be derived by differentiation of the definition equations of the four main state functions
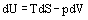



The four Maxwell Relations are important thermodynamic partial derivatives which allow the substitution of measurable properties for unmeasurable properties in thermodynamic relationships. Assuming constant composition, they can be derived from the definition equations of the basic reference properties, Equations (28) to (31):
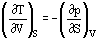
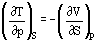
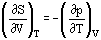

Heat capacities can also be derived from basic equations.
The heat capacity at constant pressure is given by

and the heat capacity at constant volume by

Equations for any partial derivative of thermodynamic properties in terms of measurable properties cp, (∂V/∂p)T and (∂V/∂T)P are given in the Bridgeman Tables. These relationships can then be used to construct thermodynamic diagrams. Examples for the four most common diagrams are shown in Figures 2–5.

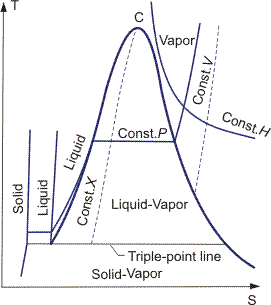
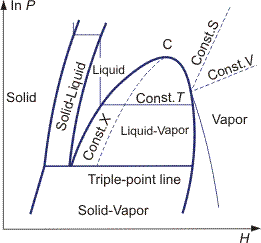
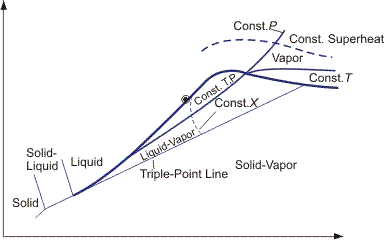
It is necessary to choose zero levels or reference states for tabulation of thermodynamic properties. In SI tables, these standard conditions are defined as a temperature of 298 K and a pressure of 0.1 MPa. Properties at standard conditions are identified by the symbol °, e.g., h°, g° etc.
For a chemical reaction

the Enthalpy of Reaction (Heat of Reaction) is defined as

If ΔHr > 0 the reaction is endothermic, if ΔHr < 0 it is exothermic. To avoid tabulation of enthalpies of reaction over a wide range of temperatures and pressures, the Standard Enthalpy of Reaction  is used in conjunction with Hess' Law
is used in conjunction with Hess' Law

where the subscript i refers to component i, and cp is molar specific heat.
The Standard Enthalpy of Reaction is usually found in tables, but can also be calculated from the Standard Enthalpy of Formation per mole of the compounds participating in the reaction

where  of single-element compounds is zero. The entropy change associated with a chemical reaction is calculated from
of single-element compounds is zero. The entropy change associated with a chemical reaction is calculated from

Most chemical reactions do not go to completion, but reach some equilibrium conditions. Consider a reaction between ideal gases:
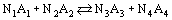
From Equation (26)

it can be derived for a reaction between ideal gases at temperature T and reference pressure 1 bar:

where 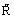 is the universal gas constant or, with the definition of the equilibrium constant Kp:
is the universal gas constant or, with the definition of the equilibrium constant Kp:

From Equations (23), (42) and (46)

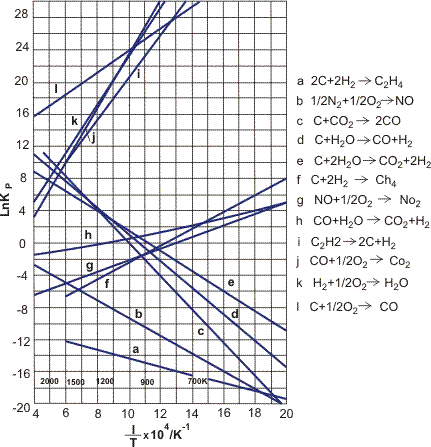
The second term on the righthand side of Eq. 47 is only moderately depending on the temperature, resulting in a straight line if the natural logarithm of Kp is plotted versus 1/T, see Figure 6. This is expressed in the van't Hoff equation

Using Dalton’s law, the equilibrium constant can be expressed in terms of mole fraction and total pressure as
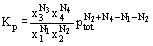
The definition of the Gibbs Energy expresses the experience that the total energy released by a process is usually different from the "useful" or "free" energy produced. This is evidence of a special demand exerted by the system or its environment, as the energy taken up by this special demand is then unavailable for useful work. In a chemical reaction, the enthalpy change ΔHr is the net energy released or taken up as a result of the making or breaking of chemical bonds. The "special demand" involves some other form of energy, due to the movement of atoms and molecules. A reaction producing more molecules from fewer creates more opportunities for molecular movement, as does producing gases from solids or more flexible molecules from more rigid molecules. These opportunities for movement constitute disorder in the system. The change in disorder is associated with the change in entropy, related to the possible energy states in the system. Where a change or reaction decreases the disorder in the system, disorder is leaving the system and its entropy change is negative. A reaction producing fewer molecules from a larger number of reacting molecules is likely to have a negative ΔS. The magnitude of ΔS is usually small and does not effect the spontaneity of the reaction at low temperatures. However, at high temperatures, the term TΔS may over-compensate the enthalpy change ΔH and reverse the direction of the reaction.
In many engineering applications, two or more phases are interacting to form equilibrium states. Mass may be exchanged between the phases, changing the concentration of components in each phase. Equilibrium is reached when the driving forces for heat transfer, mass transfer and chemical reaction have disappeared. Equation 26 written in a more generalized form is

The derivatives for constant T and p are called the Chemical Potential μi of species i in a mixture. Hence, with Equation (30)
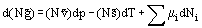
where 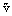 and
and  are molar volume and molar entropy.
are molar volume and molar entropy.
For two phases x and y in thermodynamic equilibrium it can be derived that

The chemical potential in an ideal gas mixture is obtained from Equation (53)

in conjunction with the definitions of entropy and Gibbs energy

Components of ideal solutions have similar molecular size and properties (for example hydrocarbon mixtures). In this case, the solution for ideal gases applies

Combining Equations (54) and (55). Raoult's Law is obtained as the simplest relationship for equilibrium between liquid and vapor mixtures

The Gibbs energy of liquids is almost independent of the pressure. For the Gibbs energy of the vapor phase at constant temperature

Hence

For equilibrium conditions, the Gibbs energies of the pure species must be identical. This yields the well-known relationship.

For a single substance at constant temperature

After substitution of enthalpy using the First Law of Thermodynamics, of specific volume using the Ideal Gas Law, and several algebraic manipulations one obtains

where° indicates a reference condition. The importance of this equation is that it relates the chemical potential and the Gibbs energy to simple measurable properties such as temperature and pressure. To extend the applicability of Equation (61) to real gases, liquids or solids, Lewis introduced the concept of Fugacity.

Reference conditions p° and y° can be selected at will, while T must be constant. Fugacity is a corrected pressure, to take into consideration interaction between atoms/molecules. For ideal gases, pressure and fugacity are identical. Since all gases and gas mixtures are ideal for low pressure

The residual Gibbs energy is the difference between the Gibbs energy calculated according to Eq. (62) and for an ideal gas
is the difference between the Gibbs energy calculated according to Eq. (62) and for an ideal gas

where the dimensionless property of the mixture φ is called the fugacity coefficient. It can be directly related to the p V T data through

where Z is the Compressibility Factor defined by Z =  . Liquid solutions are more conveniently dealt with through properties that measure their deviations from ideal solutions, rather than from ideal gases. The activity ai of a component in solution is defined as the ratio of the fugacity at a state divided by its fugacity at a reference state
. Liquid solutions are more conveniently dealt with through properties that measure their deviations from ideal solutions, rather than from ideal gases. The activity ai of a component in solution is defined as the ratio of the fugacity at a state divided by its fugacity at a reference state

Different reference states are used, depending on the actual fluid. A common standard pressure is the atmospheric pressure, i.e., 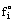 = pi for most gases. The partial excess Gibbs energy of a species in solution is then
= pi for most gases. The partial excess Gibbs energy of a species in solution is then

The activity coefficient γ

is the correction factor for the departure of a solution from ideal behavior. With the above definitions and the assumption that the fugacity in the gas phase is identical to the partial pressure, Raoult's Law can be generalized to
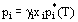
Activity coefficients depend on the concentration of the component in the solution with

which indicates that any system becomes ideal for xi → 1. Depending on the interaction between atoms/molecules, activity coefficients can be larger or smaller than 1, as shown in Figure 7 for binary mixtures of methanol and tetra-chlor-carbon, and of acetone and chloroform, respectively.

For higher pressures, nonideal behavior of the gas/vapor phase has to be considered, as well. Knowledge of activity and fugacity coefficients allows prediction of phase equilibria, solubilities and reaction rates.
References:
- Bett, K. E., Rowlinson, J. S., and Saville, G. (1992) Thermodynamics for Chemical Engineers, The Athlone Press.
- Callen, H. B. (1985) Thermodynamics and Thermostatistics, 2nd edn., John Wiley and Sons.
- Daubert, T. E. (1985) Chemical Engineering Thermodynamics, McGraw-Hill.
- Jones, J. B. and Hawkins, G. A. (1986) Engineering Thermodynamics, 2nd edn., John Wiley and Sons.
- Holman, J. P. (1988) Thermodynamics, 4th edn., McGraw-Hill.
- Howell, J. R. and Buckius, R. O. (1987) Fundamentals of Engineering Thermodynamics, McGraw-Hill.
- Rogers, G. and Mayhew, Y. (1992) Engineering Thermodynamics—Work and Heat Transfer, 4th edn., Longman Scientific and Technical.
- Smith, J. M. and Van Ness, H. C. (1987) Introduction to Chemical Engineering Thermodynamics, 4th ed., McGraw-Hill.


Comments
Post a Comment